

ALS, PLS, PMA: Namen mit einer Bedeutung?
Prof. Dr. W. ROBBERECHT
Neurologie, UZ Gasthuisberg
Leuven
Das Planen einer Bewegung entsteht in eine Serie von komplizierten Netzwerke im Gehirn, wobei die Funktion u.a. fehl schlägt wie bei der Krankheit von Parkinson und Huntington. Damit es zu einer Bewegung kommt und also Muskelkraft generiert wird, muss man zwei Arten von Nervenzellen aktivieren. Eine erste Art von Nervenzellen befindet sich im Gehirn (in der motorischen grauen Masse, Rinde oder Cortex genannt); eine zweite Art im Rückenmark um Muskeln in Arm, Bein und Rumpf zu gebrauchen, oder im Hirnstamm (der untere Teil vom Gehirn) um schluck-und Sprachmuskulatur zu gebrauchen. Da diese Nervenzellen (Neuronen genannt) verantwortlich sind für die Motorik, werden sie Motorneuronen genannt. Die sich im Gehirn befindlichen werden höhere oder Zentralneuronen genannt weil sie sich im zentralem Nervensystem befinden. Die sich im Hirnstamm oder Rückenmark befindlichen werden untere oder periphere Motor - Neuronen genannt.

Abbildung 1: die oberen Motor - Neuronen im Gehirn und
Die unteren im Hirnstamm und Rückenmark.
Die oberen Motor - Neuronen schicken elektrische Signale durch Kabel zu den unteren Motor - Neuronen. Diese Kabel sind lange Auslaufer vom Neuron die man Axonen nennt. Al diese Axonen zusammen nennt man im Medizinischen Sprachgebrauch die Corticospinale Bahn, weil diese von der Cortex bis zum Rückenmark verlauft, das im Lateinischen “ medulla spinalis” genannt wird. Die elektrischen Signale erreichen und aktivieren die unteren Motor - Neuronen. Diese letzteren schicken wiederum ihre Auslaufer oder axonen zu den Muskeln. Die axonen formen die Nerven die vom Rückenmark zu den Muskeln verlaufen. Diese axonen sind manchmal sehr lang. Wo ein Neuron nur ein Durchmesser hat von einige zehntel Millimeter, kann ein axon manchmal ein Meter lang sein (also 10.000 mal mehr) Es muss zum Beispiel vom Rückenmark bis zur Fuss-Muskel reichen (Bild 2)
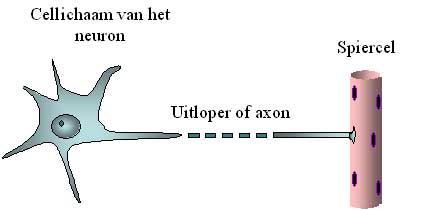
Abbildung 2: schematische Darstellung vom Zellenkörper des Motor - Neurons und sein Axon. Das Axon ist bis zu 10.000 mal länger als der Durchschnitt des Zellenkörper und ist deswegen unterbrochen dargestellt.
Während der Körperuntersuchung kann der Neurologe feststellen ob die Motor - Neuronen normal funktionieren. Mit dem EMG (Abkürzung für Elektro-Myographie) können Abweichungen der peripheren Neuronen festgestellt werden die bei der Körperuntersuchung noch nicht feststellbar sind.
Es sind diese zwei Neuronen die schlecht funktionieren und später schmachten (degenerieren) in der motor – Neuronen - Krankheit oder motor – Neuron - Degeneration. Von diese Art von Krankheiten ist die Amyotrophe Lateral Sklerose die am meisten bekannte, aber es gibt verschiedene Variationen. Schlechtes funktionieren der höheren Motor - Neuronen gibt Anlass zu Krämpfen und anormal lebendige Reflexe; Ausfall von niedrigen Motor - Neuronen ergibt Muskelschwäche und Muskel-Atrophie. Dieses letzte Wort bedeutet das abmagern einer Muskel was man sehr gut feststellen kann an der Hand.
Bei Amyotrophe Lateral Sklerose (ALS) sind es die beiden Arten von Neuronen (sowohl die Zentralen wie die Peripheren) die erkranken und absterben. Dadurch entsteht eine Kombination von Anzeichen: Muskelschwund durch Degeneration der peripheren Neuronen und zu gleicher Zeit anormale Reflexe und Krämpfe die hin weisen auf Degeneration der Zentral - Neuronen. Die Kombination zusammen mit dem stets stärker werdenden Krankheitsbild, erlauben dem Arzt ALS fest zu stellen. Das EMG wird den Anfall auf den Peripheren Neuronen bestätigen.
In den Medien werden ALS und die andere Motor - Neuronen – Krankheiten öfter als Muskelkrankheiten umschrieben. Dies ist leicht zu verstehen ,da die schlechte Funktion der Muskeln am meisten sichtbar ist. Es sind jedoch nicht die Muskeln die „Fehlerhaft“ arbeiten, sondern die Nervenzellen die für die nötigen Signale sorgen müssen.
ALS kommt vor bei ungefähr 1 bis 2 Menschen pro 100.00 pro Jahr. Es gibt nahezu 800 ALS-Patienten in Flandern. ALS kommt mehr vor bei Männer als bei Frauen. Die meisten ALS-Patienten (90%) haben keine Familienmitglieder mit derselben Krankheit: wir nennen es sporadische oder nicht Familiäre Fälle. Ungefähr 10% der Patienten haben allerdings Familienmitglieder mit ALS in der familiären Form. Von diese Art kennen wir in 20% der Fälle die Krankheitsursache (Anomalien des SOD1 Gen)
Bei so manchen Patienten beschränkt sich die Krankheit im Anfangsstadium zu den unteren Neuronen. Diese Menschen entwickeln Muskelatrophie und Kraftverlust, aber keine Krämpfe. Bei diesen Patienten entwickelt sich dann aber sehr schnell Krankheitszeichen des höheren Neurons. Es handelt sich dann jedoch um ALS. Ein anderer Teil der Patienten entwickeln aber nie Krankheitszeichen der zentralen Motor - Neuron-Krankheit. Neurologen haben keinen richtigen Namen für diese Krankheit. Es wird Untere-Motor - Neuronen-Krankheit genannt oderProgressive Spino-Muskuläre Atrophie (PSMA) oder Progressive Muskuläre Atrophie. Diese Krankheit neigt zu langsameren Verlauf als ALS, aber man weiss es nicht richtig. Es ist sehr gut möglich dass die Form eine Variante von ALS darstellt: manche Menschen mit der SOD1 Mutation entwickeln nie Krämpfe und haben per Definition doch ALS.
Ein kleiner Teil der Patienten entwickelt einige Abweichungen der unteren Motor - Neuronen in den Armen und einige Abweichungen der höheren Motor - Neuronen in den Beinen. Auf Englisch nennt man das „ flail arm Syndrome“. Man weiss dass diese Krankheit langsam fortschreitet. Nur ganz spät in der Evolution dieser Form entstehen Probleme mit der schluck- und Sprachmuskulatur.
Das Umgekerhte gibt es auch. Bei manchen Patienten sind nur die zentralen Motor - Neuronen befallen. Diese Form der motor - Neuronen-Krankheit nennt man Primäre Lateral Sklerose (PLS). Jedoch nach Verlauf der Zeit wird bei einem Teil der Patienten deutlich dass es sich um Schädigung der unteren Motor - Neuronen handelt; diese Menschen haben dann letztendlich auch ALS. Um die Evolution in Richtung ALS nach zu gehen, wird bei diesen PLS-Patienten regelmassig ein EMG durchgeführt.
Ein anderer Teil der Patienten entwickelt nie eine Evolution Richtung ALS. Sie haben wirklich PLS. Über diese Krankheit ist nicht viel bekannt. Wir wissen nur dass diese Krankheitsform zehnmal weniger vorkommt als ALS. Schätzungsweise gibt es in Flandern ungefähr 50 Fälle. Es ist jedoch wichtig eine korrekte Diagnose zu erstellen damit der Unterschied zu ALS gemacht werden kann. Die Aussichten für PLS Patienten sind nämlich verschieden: die Krankheit evoluiert viel langsamer als ALS. Eine Krankheitsdauer von 10 oder sogar 15 Jahre ist nicht aussergewöhnlich für PLS aber schon für ALS.
Die Krankenversicherung zahlt riluzole nur dann zurück wenn an alle ALS Kriterien Folge gegeben ist. Anfangs erfüllen viele ALS-Patienten diese Bedingungen nicht, und würden diese Medikation nicht zurück erstattet bekommen. Im Grunde genommen bedeutet dies Zeitverlust: erst nach einigen Monaten oder ein Jahr zeigen sich die Krankheitsbilder die vom RIZIV erfordert werden. Jedoch meinen viele ALS-Spezialisten dass riluzole gerade in der Anfangsfase sehr viel Zweck hat.
Weil das RIZIV aber rizuole nicht zurück bezahlt Für Menschen mit Abweichungen an den unteren Motor - Neuronen ist bedauerlich weil diese Patienten wie gesagt fast immer an eine form von ALS leiden. Auch für PLS zahlt die Krankenversicherung nichts zurück. Das letzte ist verständlich, da die Evolution dieser Krankheit so langsam fortschreitet das unterstellt werden kann das kein Nützen zu erwarten ist. Ganz sicher weiss man es aber nicht. In den Niederlanden hat man eine Studie angefangen um dies nach zu gehen. Das Resultat ist vorhersehbar.