

Est-ce que la SLA se diffuse et se propage?
13-12-2012
Auteur: John M. Ravits, M.D. Professor of Clinical Neuroscience, Head ALS and Neuromuscular Translational Research University of California, San Diego
Parmi les mystères de la SLA il y a la différence de symptômes d'un patient à l'autre. Chaque patient semble faire face à un ensemble différent de problèmes et de défis. Certains ont des difficultés à parler, certains à marcher, certains avec leurs bras, d’autres avec leur tronc. Il est même reconnu que certains patients ont des problèmes de langage, de comportement ou de personnalité. Il suffit d’assister à une collecte de fonds, un groupe de soutien, une réunion ou de consulter un site Web pour le constater.
Comment cela est-il possible? Comment une maladie peut-elle différer apparemment tellement d'une personne à l'autre ? Quel est le tronc commun qui permet le même diagnostic ?
L'expression médicale pour ces différentes apparences est l’hétérogénéité du phénotype. Les phénotypes sont l'apparence des choses. L'hétérogénéité est la différence. Dans la SLA, l’hétérogénéité du phénotype inclut l'emplacement de la faiblesse — par exemple, le langage et la déglutition (souvent appelé bulbaire) ou les membres— et le caractère de la faiblesse, motoneurones supérieurs causant rigidité et spasticité ou motoneurones inférieurs causant faiblesse et atrophie —ou les deux.
Derrière la faiblesse il y a la dégénérescence ou la disparition de motoneurones du système nerveux central. Ce processus de dégénérescence sélectionne les motoneurones avant tout autre comme cible. Ainsi, ce qui est commun à tous les patients est un problème sous-jacent de dégénérescence de motoneurones, situé n'importe où dans le système nerveux central et causant n’importe quel problème. La caractéristique fondamentale de la SLA est la dégénérescence des motoneurones et la perte de motricité en découlant.
Comment cela explique-t-il l'hétérogénéité du phénotype ? Il s'avère que les motoneurones résident en des endroits anatomiques précis dans le cerveau, le tronc cérébral et la moelle épinière. Leur emplacement dans le système nerveux détermine quels muscles du corps ils contrôlent et la manière dont ils les contrôlent. Le système moteur est donc un réseau de circuits électriques pour les muscles. Il a fallu un effort titanesque aux neurologues cliniciens, neuroanatomistes, neuropathologistes et neurophysiologistes en Allemagne, Autriche, Russie, Angleterre, Australie, Italie, Espagne et France et plus de 50 ans de travail dans la seconde moitié du XIXe siècle pour comprendre les schémas de connections. Et ces découvertes ont eu lieu en même temps que la SLA, maladie qui affecte cet ensemble, a été identifiée et comprise. L'élucidation simultanée des deux ––le système moteur et la maladie qui l’agresse––, a clairement établi comment ils peuvent être liés ensemble.
Récemment, des recherches de certains neurologues dans plusieurs pays ont soulevé des questions à ce sujet, comment les deux pourraient être intimement liés. Au début de la maladie, avant les choses ne deviennent trop compliquées, les neurologues cliniques peuvent supposer et confirmer par simple examen clinique l’endroit où les motoneurones défectueux sont situés dans le système nerveux. Ils peuvent localiser les problèmes dans les réseaux. Deux caractéristiques importantes sont maintenant de plus en plus envisagés: 1) la SLA commence de façon très localisée presque n'importe où dans le circuit moteur, peut-être même aléatoirement ; et 2) la SLA semble progresser vers l'extérieur de cet endroit le long de voies anatomiques, la région adjacente ou au sein de réseaux spécifiques.
Seulement, cela implique que la SLA se propage ! Il peut être localisé au début, touchant des régions distinctes du cerveau ou de la moelle épinière, mais ensuite il avance ou se déplace anatomiquement en recrutant ou capturant d’une certaine façon des motoneurones dans sa proximité immédiate. Ainsi, l'hétérogénéité surprenante du phénotype peut s'expliquer par 1) le site spécifique, peut-être aléatoire, où elle commence dans le système nerveux; 2) la mesure très variable dans laquelle les motoneurones supérieurs et inférieurs sont impliqués et 3) la voie de propagation vers l’extérieur et cela dépend de son emplacement exact.
Depuis cinq ou dix ans, des hypothèses sur la propagation ont été évoquées dans le domaine de la neurodégénérescence. Les maladies d'Alzheimer et de Parkinson, par exemple, sont deux maladies dégénératives qui partagent avec la SLA le fait que certaines populations de neurones sont sélectivement vulnérables. Pour Alzheimer et Parkinson, la propagation est supposée se produire selon un modèle stéréotypé du début à la fin de la maladie. La SLA peut être différente parce qu'elle semble se déclarer au hasard à un endroit du système moteur, soit parce qu'elle est réellement différente ou parce que cette manière de propagation ne peut pas facilement être observée dans les autres maladies. Les maladies de Parkinson et d'Alzheimer sont-elles différentes à cet égard ? Ou le SLA nous montre-t-elle une facette spéciale de la neurodégénérescence ? Ce sont les pièces du puzzle qui sont actuellement discutées et étudiées.
La première question concernant la SLA est : est-ce que cela se passe réellement et de façon ordonnée ? Ou y a-t-il d'autres explications, telles que les programmations génétiques ou développementales, qui pourraient mieux expliquer le déclenchement et la progression de la SLA chez chaque patient ? Un indice se trouve dans la SLA familiale (fSLA). Un défaut génétique très spécifique est la cause de cette maladie. Cette mutation génétique affecte chaque cellule du corps, pas seulement un emplacement ou un type de cellule. Il est clair qu'il y a une hétérogénéité marquée de phénotype au sein de la famille, sans doute semblable à l'hétérogénéité qui se produit dans la SLA sporadique. Certains membres de la famille développent la forme bulbaire de la maladie, d'autres commencent dans les bras ou les jambes et dans la plupart des cas, la maladie se propage vers l'extérieur. Ainsi, le défaut génétique provoque la déclaration de la maladie à un endroit anatomique donné et ceci à son tour en détermine le phénotype.
Comment la propagation pourrait-elle se produire au niveau cellulaire et moléculaire ? Il y a beaucoup de possibilités différentes. Une possibilité est que le microenvironnement cellulaire des neurones soit corrompu par la maladie et que ces variations locales induisent une cascade d'événements qui se propageraient vers l'extérieur. Il s’avère que d’autres cellules sont impliquées dans ce domaine, comme les astrocytes et les cellules de la microglie, ainsi que les oligodendrocytes, ces cellules assurant des fonctions vitales distinctes de celles des neurones. Une autre façon qui a émergé est qu’un (des) facteur(s) toxique(s) soluble(s) se diffuserai(en)t à travers le système nerveux, provoquant un déversement toxique diffusé vers l'extérieur. Et pendant de nombreuses années des propriétés importantes de pliage des protéines ont été étudiées largement dans ce que l'on appelle la biologie des prions, la propriété-clé étant qu'une protéine peut se replier de façon défectueuse et que cette protéine mal repliée peut en induire la même défectuosité chez d’autres et ainsi se diffuser dans ce qui est appelé une propagation du type prion.
La propagation a d'importantes implications thérapeutiques ! Tout d'abord, si nous pouvions comprendre le processus de propagation lui-même, nous pourrions le prendre comme cible thérapeutique, combattant ainsi le processus qui attaque la cellule.
Ensuite, si nous pourrions commencer au début de la maladie, nous pourrions mener ce combat dans une région limitée. Les médicaments peuvent atteindre toutes les régions où ils sont nécessaires. Mais les stratégies régionales sont différentes : elles sont locales. La différence est similaire à l'utilisation d'un antibiotique systémique ou d’une pommade locale pour traiter une infection. Cette différence n’est nulle part plus importante que dans les traitements actuels avec des cellules-souches. Pour gagner une lutte thérapeutique par cellules-souches, elles doivent être placées dans des régions stratégiques du système nerveux. Elles ne peuvent pas être administrées systémiquement. Mais quels sont les « régions stratégiques » ? Sont-elles les régions où la maladie est le plus active ? Ou sont-elles les régions où la pathologie progresse mais pas encore affectées (dans le but de défendre ceux-ci en circoncisant les autres) ? Cela devra être défini à l'avance selon le progrès de ces thérapies.
Au cours des prochaines années, la propagation de la maladie sera étudiée de plus près et nous mènera certainement à de nouvelles idées, nous rapprochant de l'objectif global d'une thérapie fondamentale directe contre la SLA.
Image
Un modèle de propagation de la SLA sur base d’un foyer limité et d’une diffusion.
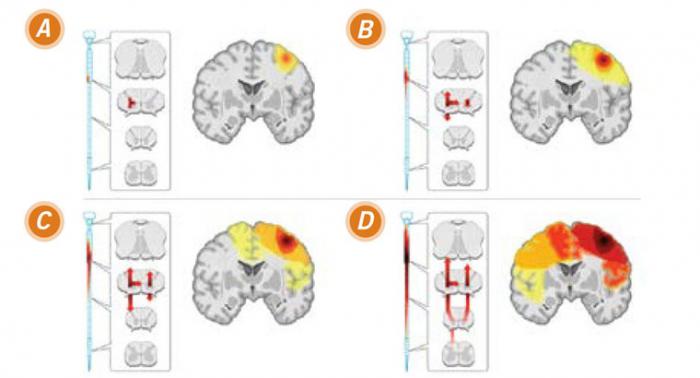
Image courtesy of Ravits, J. M. et al. Neurology 2009;73:805-811
(A) début : au début de l’affectation clinique, la dégénérescence implique des neurones dans le cerveau et la moelle épinière qui innervent la même région périphérique du corps (ici l'innervation de la main droite).
(B) début de la propagation : au fil du temps, le processus de la maladie se propage dans le cerveau et la moelle épinière et les manifestations cliniques augmentent.
(C) continuation de la diffusion vers l'extérieur : avec le temps, la maladie devient de plus en plus complexe.
(D) propagation maximale : en fin de compte, le processus de la maladie semble être diffus et symétrique, les détails dépendent de l'endroit de l’apparition de la maladie.
Traduction : Fabien
Source : Research ALS Today