

De nouveaux progrès dans la pathogénie et la thérapeutique de la sclérose latérale amyotrophique
11-04-2021
La sclérose latérale (SLA) est une maladie neurodégénérative handicapante avec une incidence de 1 - 5 cas/100 000 sujets. Elle se caractérise par une dénervation, une faiblesse, une atrophie et une paralysie musculaire, entraînant le plus souvent le décès du patient (par insuffisance respiratoire) dans les 3 - 5 ans suivant le diagnostic. La maladie se manifeste par une perte progressive des motoneurones supérieurs et inférieurs liée à une étiologie complexe (gènes et facteurs environnementaux). Les formes sporadiques sont les formes les plus communes (jusqu’à 90 % des cas), mais il est difficile de les distinguer cliniquement et histopathologiquement des autres formes connues jusqu’à présent. Ces formes sont dues à des mutations de plus de 25 gènes liés à la SLA dont (i) le gène codant pour la superoxyde dismutase‐1 (SOD1), une enzyme clé antioxydante, (ii) les TARDBP et FUS codant pour les protéines TAR ‐DNA binding portein‐43 (TDP‐43) et fused in sarcoma (FUS) respectivement responsables de l’épissage, le transfert et la stabilité du pré‐ARNm et (iii) le C9orf72 codant pour la protéine responsable du trafic intracellulaire dans les neurones et d'autres fonctions cellulaires encore peu connues. Des changements dans les SOD‐1, TARDBP, FUS et C9orf72 sont observés dans la majorité des cas (environ 70 %) de SLA familiale (Kim, Gautier, Tassoni‐Tsuchida, Ma, & Gitler, 2020).
SOD-1 est le premier gène lié à la SLA identifié en 1993 (Rosen et al.,1993), lors du développement de modèles animaux transgéniques (principalement des souris) qui ont présenté les mutations de SOD-1 observées chez des patients (la plus fréquente est G93A ; Ripps, Huntley, Hof, Morrison, & Gordon, 1995). Pendant des années, cette méthode a été le seul outil pour l'étude de la pathogénie et de la thérapeutique de la SLA. La Figure 1 montre la progression importante des nombres d’articles publiés relatifs aux études menées sur la SLA et collectés par PubMed au cours des 50 dernières années. Comme le montre la Figure 1, les découvertes de nouveaux gènes liés à la SLA (TARDBP, FUS et C9orf72) ont fourni des modèles transgéniques surexprimant les mutations les plus fréquentes de ces gènes et ont, ainsi, contribué au progrès de la recherche sur la SLA (Van Damme, Robberecht, & Van Den Bosch, 2017).
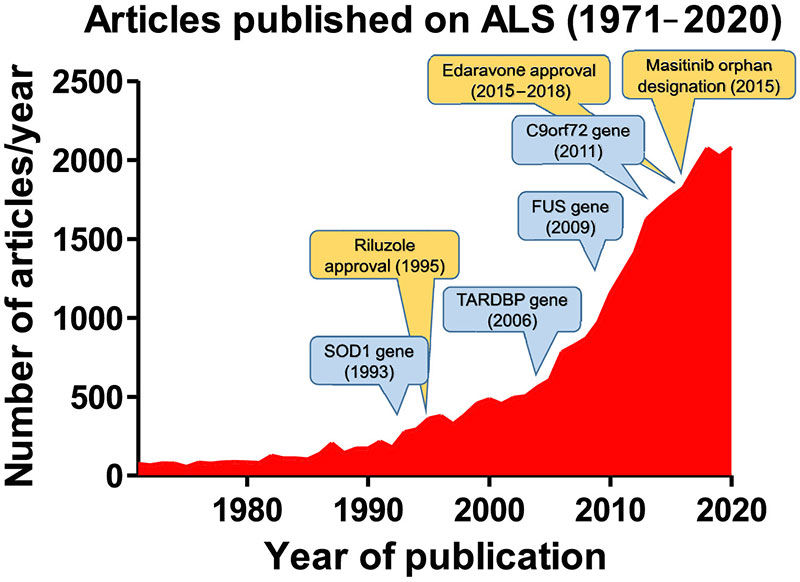
FIGURE 1
L’évolution des articles sur la SLA publiés au cours des 50 dernières années selon une étude menée par PubMed avec le terme “amyotrophic lateral sclerosis” et mention de l'année de publication et de découverte des gènes les plus pertinents liés à la SLA et de l'approbation des quelques médicaments disponibles pour les patients atteints de SLA.
Comme mentionné plus haut, il est difficile de distinguer cliniquement les formes sporadiques et familiales de la SLA. Il en va de même pour les mécanismes pathogéniques de la mort des motoneurones. La découverte progressive des différents gènes liés à la SLA et le développement de modèles expérimentaux basés sur ces gènes ont permis une meilleure compréhension de ces mécanismes pathogènes, comme l'indique la Figure 2.

FIGURE 2
Le diagramme montre les différents mécanismes pathogéniques responsables de la dégénérescence des motoneurones et leur rapport avec certains gènes liés à la SLA.
La SLA reste une maladie incurable. Les traitements actuellement disponibles ne prolongent la durée de vie que de quelques mois, d’où le besoin majeur de médicaments plus efficaces. Parmi ces agents thérapeutiques, nous citons le riluzole (Rilutek®), agent anti‐excitotoxique, autorisé pour la première fois en 1995 et qui était pendant longtemps le seul médicament disponible pour les patients atteints de SLA. En 2015, l’edaravone (Radicava®), agent antioxydant, a été approuvé au Japon, puis aux États-Unis (2017) et au Canada (2018). La même année, le masitinib (Kinavet‐CA1®), un anti‐inflammatoire et un inhibiteur de protéines tyrosine kinases a été désigné comme un médicament orphelin dans le traitement de la SLA. Ce sont tous les médicaments modificateurs de la maladie disponibles pour les patients atteints de SLA. Toutefois, au cours des années, plusieurs agents thérapeutiques avec différents mécanismes d’action (anti‐excitotoxiques, antioxydants, anti‐inflammatoires, agissant sur les mitochondries, facteurs neurotrophiques et autres agents) ont été testés. Bien que ces agents aient présenté un bon profil de sécurité et d'efficacité sur les modèles animaux, ils n'ont pas pu avoir les mêmes effets lors des essais cliniques (la Figure 3 récapitule les agents thérapeutiques les plus efficaces évalués au cours des dernières années). Plusieurs facteurs sont considérés comme des raisons possibles des échecs comme la prévisibilité des modèles expérimentaux, le démarrage tardif des essais cliniques, les mécanismes pathogéniques hétérogènes de la SLA ou le besoin de stratégies multi‐cibles.
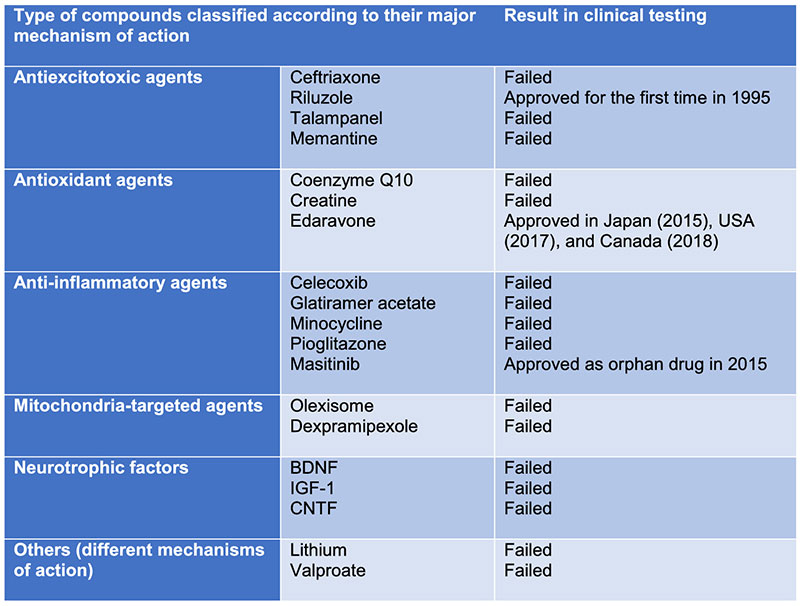
FIGURE 3
Ci-dessus un tableau des familles de composés étudiées pour leurs propriétés neuroprotectrices (modificatrices de maladie) dans la SLA et les familles de composés ayant fait l’objet d’essais cliniques avec les résultats obtenus (adapté de Fernández-Ruiz, 2019).
L’identification des nouveaux gènes liés à la SLA lors des 15 dernières années a fourni des informations clés relatives aux mécanismes moléculaires et cellulaires déclenchant la pathologie ou contribuant à la progression de la maladie. Ces découvertes ont aidé à identifier de nouvelles cibles thérapeutiques en vue de développer des médicaments neuroprotecteurs. De plus, ces nouveaux gènes ont fait le lien entre la SLA et d'autres pathologies neurodégénératives comme la démence fronto-temporale (DFT). Ces deux pathologies sont aujourd’hui considérées comme deux aspects de la même pathologie. L’objectif de ce numéro thématique est de faire le point sur les progrès récents dans la SLA et des pathologies associées, afin de fournir à la communauté scientifique une vision actualisée de l'étiologie de la SLA, des mécanismes pathogéniques impliqués dans la dégénérescence des motoneurones et du développement actuel de traitements nouveaux et efficaces.
Ce numéro thématique est extrait du “International Workshop on ALS: new genes, new treatments, new hopes” organisé le 30 et 31 octobre 2019 à Madrid, avec le support du BJP.La première partie est un article de Javier Riancho, Adolfo López de Munaín et leurs collègues traitant d'une nouvelle conceptualisation de l'étiologie de la SLA. Ces auteurs ont décrit la SLA comme une maladie complexe causée par plusieurs facteurs : risque génétique individuel, vieillissement et facteurs environnementaux qui déterminent l’apparition, la progression et le pronostic de la maladie. Ils débattent de l’impact des facteurs environnementaux modifiables et passent en revue les données relatives au cancer, à l'auto-immunité et aux troubles métaboliques, en tant que causes possibles de la SLA (Riancho et al., 2020).
Dans le deuxième article, Riancho et ses collègues ont abordé la SLA sous l'angle d'une maladie touchant non seulement le système moteur, mais aussi le système sensoriel. Ils ont analysé les informations fournies par les patients et les modèles animaux et ont mis en évidence l'altération des réseaux sensorimoteurs comme un signe important de cette maladie, souvent ignoré ou sous-estimé (Riancho, Paz‐Fajardo et López de Munaín, 2020).
Dans l’article suivant, Rosario Osta et ses collègues ont rassemblé les données expérimentales recueillies dans plusieurs laboratoires relatives à la contribution importante des déficiences dans les tissus périphériques, en se concentrant sur les muscles squelettiques. Ils ont évalué le rôle de plusieurs facteurs comme le dysfonctionnement des mitochondries dans les muscles, le métabolisme énergétique, la protéostase et le métabolisme de l'ARN, ainsi que les défauts de la myogenèse dans l'apparition de la maladie. La plupart des informations recueillies par ces auteurs dans l’article ont été obtenues chez les modèles de souris mutantes SOD‐1, le modèle classique de SLA basé sur le premier gène lié à la SLA identifié. Ils ont également évoqué les efforts mis en œuvre pour développer de nouvelles stratégies thérapeutiques ciblant le muscle squelettique afin de ralentir l'apparition et la progression de cette maladie (Manzano et al., 2020).
La ribonucléoprotéine TDP‐43 découverte dans la SLA en 2006 est très souvent observée dans cette pathologie (et aussi dans la DFT), sous forme d'agrégats dans le cytosol, et ce, non seulement dans les cas familiaux issus de mutations du gène TARDBP, mais aussi dans plusieurs cas sporadiques (jusqu'à 95 % des cas de SLA). Ceci est dû aux modifications post-traductionnelles (par exemple, l'ubiquitination, la phosphorylation, l'acétylation, la sumoylation et le clivage). Ces modifications se produisent dans le TDP‐43 et entraînent une modification de sa conformation et de sa localisation, une dérégulation, une altération de sa fonction, un dépôt et une agrégation. Emanuele Buratti a étudié en détail ces phénomènes dans le cadre de sa participation à ce numéro thématique. Il a souligné le potentiel thérapeutique de plusieurs composés de faible poids moléculaire, capables d’agir sur les caractéristiques pathologiques du TDP‐43, notamment les niveaux d’expression, la mauvaise localisation cytoplasmique, les modifications post-traductionnelles, le clivage, la formation des granules de stress et autres (Buratti, 2020).
Dans le même sens que Buratti, Ana Martinez et ses collègues ont montré que l’inhibition de plusieurs kinases responsables de la phosphorylation du TDP‐43 et d’autres protéines liées à la SLA pourrait être une thérapie potentielle de la SLA. L'accent a été mis sur c-kit (une tyrosine kinase inhibée par le masitinib, actuellement désigné comme médicament orphelin pour le traitement de la SLA), ROCK (p. ex, le fadusil), mTOR (p. ex, la rapamycine), GSK-3 (p. ex, le lithium) et d'autres inhibiteurs déjà proposés dans le scénario clinique. L’article aborde également d’autres inhibiteurs qui se sont révélés efficaces et sûrs dans des études précliniques (comme les inhibiteurs de CK‐1, CDC7 et MAP3K) et même dans les premières phases de la découverte du médicament comme les inhibiteurs des tau-tubuline kinases (TTBKs) et des kinases dépendantes des cyclines (CDKs) (Palomo, Nozal, Rojas‐Prats, Gil, & Martinez, 2020).
Les récepteurs Sigma‐1 sont codés par le gène SIGMAR1, qui a également été lié au développement de la SLA. Parmi leurs différents substrats de cellules dans le SNC, les récepteurs sigma‐1 se trouvent généralement dans les motoneurones et sont situés au niveau des complexes intracellulaires formés par le réticulum endoplasmique et les mitochondries. Ils agissent comme des protéines chaperonnes et assurent la survie des motoneurones. Ainsi, cibler ces récepteurs pourrait être un moyen efficace de réduire la mort de ces neurones. Xavier Navarro et ses collègues ont étudié cette possibilité dans leur article, dont l’objectif est de valider les propriétés neuroprotectrices des ligands de ces récepteurs.(Herrando‐Grabulosa, Gaja‐Capdevila, Vela, & Navarro, 2020).
La SLA est également liée à des marqueurs épigénétiques, une nouvelle piste à explorer pour les chercheurs. Ludo Van den Bosch et ses collègues ont étudié la possibilité d'inhiber les histones désacétylases, une catégorie d'enzymes épigénétiques. Ils ont examiné le potentiel d'inhibiteurs connus tels que le valproate, le lithium et le resvératrol et de nouveaux inhibiteurs synthétiques des différents sous-types d'histone désacétylases notamment les sirtuines, dans des études précliniques et cliniques. Ils se sont particulièrement intéressés à la capacité des inhibiteurs de traverser la barrière hémato-encéphalique, leur tolérabilité et leur sécurité (Klingl, Pakravan, & Van Den Bosch, 2020).
Nous espérons que ces contributions seront à même de mobiliser les ressources et les efforts nécessaires à identifier de nouveaux cibles et agents thérapeutiques et d'encourager la recherche continue de médicaments efficaces pour ralentir la progression de la SLA. Les patients ont urgemment besoin de ces efforts.
Traduction : Nessrine Maiza
Source : British Journal of Pharmacology